ポスト
ポスト


月刊カレントテラピー
1年前
【解説】再生不良性貧血におけるクローン性造血 (牧島秀樹先生)

再生不良性貧血におけるクローン性造血
牧島秀樹先生 (信州大学医学部血液・腫瘍内科学教授)
再生不良性貧血におけるクローン性造血.pdf378.41KB
再生不良性貧血 (AA) は、 造血幹細胞が自己のTリンパ球に傷害されることにより発症する自己免疫性の骨髄不全症候群である。 近年の詳細なゲノム解析の結果、 AAにおける低形成骨髄は腫瘍性病変ではないものの、 クローナルな細胞集団により構成されていることが明らかとなった。 こうしたAAに伴うクローン性造血は、 古典的なX染色体のskewingにより示唆され、 PNH血球の検出、 さらにはコピー数解析によるUPD6pの発見および次世代シーケンスによる遺伝子変異の検出により証明されてきた。 AAは骨髄異形成症候群 (MDS) へしばしば移行するが、 AAとMDSのゲノム異常のランドスケープは異なる部分が多い。 AAでは、 血液疾患のない高齢者に認められるクローン造血 (ARCH/CHIP) と共通性はあるものの、 やはり異なったゲノム異常が認められる。 総じてこれらの結果は、 病因である自己免疫反応からのエスケープが、 AAにおけるクローン性造血の形成に関与することを示唆している。
Ⅰ. はじめに
再生不良性貧血 (aplastic anemia : AA) は、 自己免疫機序による造血幹細胞の傷害に起因する骨髄不全症候群の一病型である。 経過中、 遺伝子変異あるいはコピー数異常を伴うクローン性造血を認めることがあり、 発作性夜間血色素尿症 (paroxysmal nocturnal hemoglobinuria : PNH) の合併、 さらには骨髄異形成症候群 (myelodysplastic syndromes : MDS) や急性骨髄性白血病 (acute myeloid leukemia : AML) など骨髄腫瘍疾患への移行をしばしば認める。
これらの造血細胞クローンは、 AAの発症や臨床経過と深く関連するが、 AAに合併するMDS/AMLの起源に関する研究により、 また新たな知見が報告されている。 ひとつの可能性は、 自己免疫学的な造血抑制からのエスケープであり、 またひとつは、 ドライバー遺伝子変異の獲得である。
特に、 2014~2015年にかけて複数の施設より報告されたAAにおける遺伝子変異は、 従来MDS/AMLにおいて高頻度に認められ骨髄腫瘍のドライバーとみなされている変異とオーバーラップし、 陽性例では骨髄腫瘍への移行を高頻度に認める。
さらには、 AAで認められる変異の一部は、 健常高齢者において認められるクローン性造血 (age related clonal hematopoiesis : ARCH/clonal hematopoiesis of indeterminate potential : CHIP) とも共通しており、 それらもやはりオーバーラップする。 本稿では、 ゲノムワイドの検索により明らかとなった、 AAにおけるクローン性造血に関して概説する。
Ⅱ. 再生不良性貧血における自己免疫性造血不全
AAは免疫学的機序による造血幹細胞ないし前駆細胞の傷害により多系統の血球減少を呈する骨髄不全症候群であり、 細胞傷害性フェノタイプを有するT細胞の異常や疾患特異的なHLAタイプを認める¹⁾²⁾。 AAの病態における自己免疫機序の関与については、 現在、 自己反応性の細胞傷害性T細胞 (cytotoxic T lymphocyte : CTL) によって、 造血幹細胞ないし前駆細胞が傷害される結果、 骨髄の著しい低形成が惹起されるとする仮説が広く支持されている³⁾⁴⁾。
その根拠としては、 同種造血幹細胞移植の生着不全の後、 自己造血の回復が認められたとする報告や⁵⁾、 抗胸腺細胞グロブリン (ATG) とシクロスポリンの併用や大量シクロホスファミドによる免疫抑制療法が本症で高い奏効率を示すことが挙げられる⁶⁾⁷⁾。 また、 骨髄の低形成には、 免疫応答反応によって免疫担当細胞から大量に分泌されるTNFα、 TGF-β、 IFN- γなどの炎症性サイトカインによる造血抑制も骨髄不全の病態に重要な役割を担っていると考えられる⁸⁾~¹⁰⁾。
これに関連し、 われわれはAAにおけるT細胞クローンにはSTAT3の機能獲得型の変異が検出されることを見出した¹¹⁾。 この変異が、 赤芽球癆の原因となるLGL leukemiaにおいて高頻度に認められることは興味深い¹²⁾¹³⁾。
Ⅲ. 再生不良性貧血に認められるクローン性造血の存在
従来、 重症のAA症例の予後は極めて不良で、 同種造血幹細胞移植が唯一の根治的な治療手段であったが、 1980~1990年代にかけて免疫抑制療法が確立され、 幹細胞移植が適応とならない高齢者を中心として、 その治療成績には劇的な改善が認められた⁶⁾⁷⁾。 一方、 免疫抑制療法により改善した長期生存例を中心として、 AA症例のおよそ10%において、 MDS/AMLなどの骨髄系腫瘍が続発し、 免疫抑制療法後の血球減少の再燃とならび、 AAの治療上の問題点の一つとなっている¹⁴⁾~¹⁶⁾。
また、 AAには、 高頻度にPNH陽性クローンが認められ、 PNHの合併例やPNHへの病型の移行が認められる。 AAと低形成性MDSとの異同やAML・PNHへの移行や合併については、 臨床的に長年にわたって議論されており¹⁷⁾、 これらAAに関連した造血器疾患の問題は、 近年、 クローン性造血という観点から非常に注目されている (図1)¹⁸⁾~²¹⁾。
図1 再生不良性貧血から骨髄異形成症候群へ進展する際に獲得されるゲノム異常
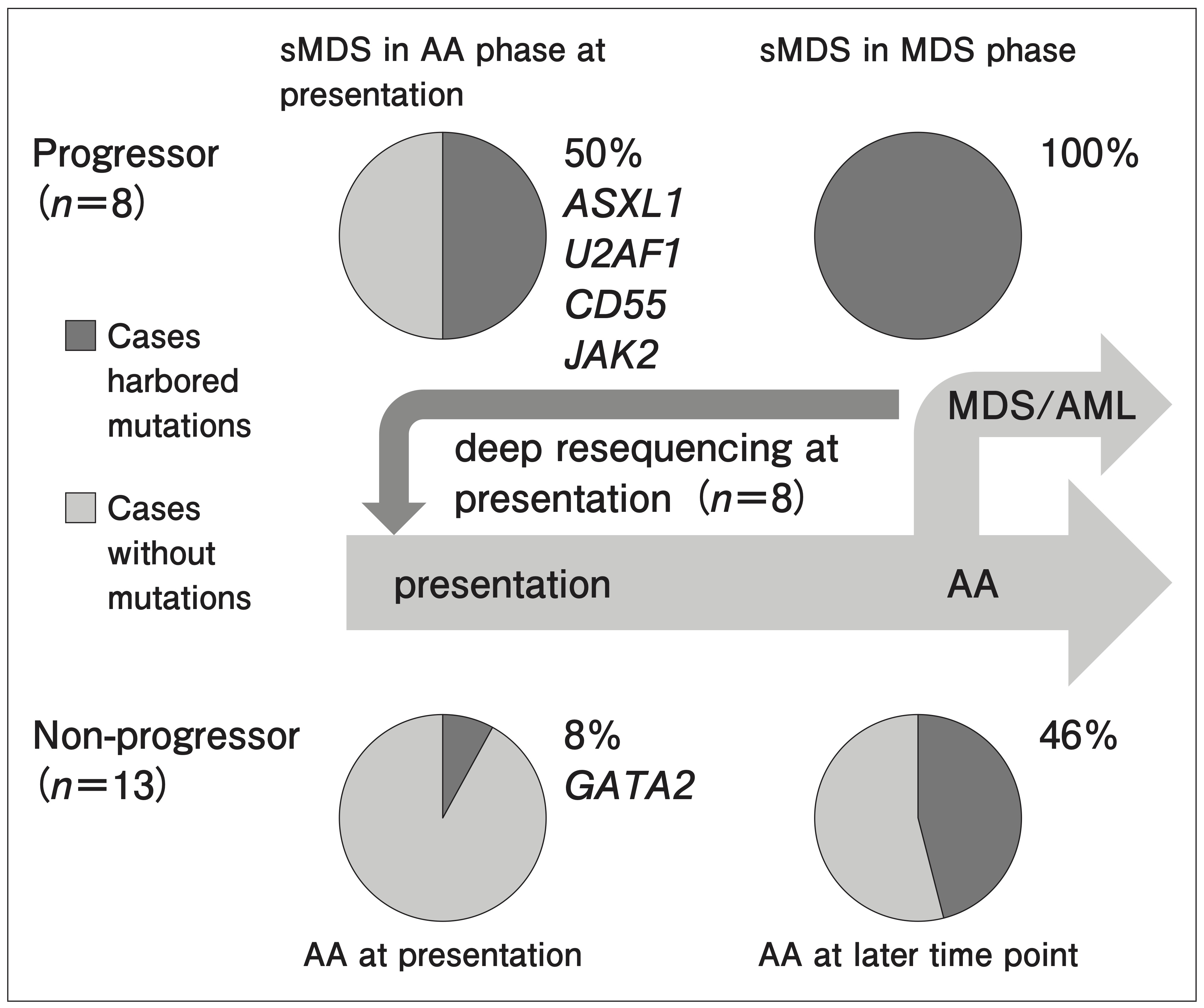
再生不良性貧血の経過中に骨髄異形成症候群へ進展した症例 (n=8) と進展しなかった症例 (n=13) におけるシーケンス解析。 sMDS : secondary myelodysplastic syndromes, AA : aplastic anemia
〔参考文献21) より引用〕
AAにおけるクローン性造血の存在については、 複数の報告により、 そのエビデンスが示されてきた。 たとえば、 本症の10~15%内外の症例で、 染色体異常が認められること²²⁾、 ランダムに生ずるX染色体の不活化にskewingが認められること¹⁸⁾、 また本症が、 経過中にPIGA 変異を有するクローンによる造血で特徴づけられるPNHに移行することから¹⁹⁾²³⁾ 以前より示唆されてきた。
AAに伴い繰り返し認められる染色体異常として、 +8や-7、 del (20q) といった、 骨髄系腫瘍でも頻繁に認められる代表的な異常が含まれる。 しかし、 AAのクローン性造血において最も高頻度に認められるのは、 6番染色体短腕に生じるコピー数異常を伴わないLOH (loss of heterozygosity) である (後述)。
Ⅳ. 再生不良性貧血におけるゲノムワイドのコピー数解析
近年、 高解像度の染色体コピー数解析法としてsingle nucleotide polymorphism array (SNP-A) が研究目的で利用されている。 本邦では外注検査により依頼でき、 異常を有するクローンが検査サンプルのおよそ20%以上を占めていれば検出可能である。 SNP-Aによるコピー数解析は、 G分染法により検出感度以下の狭小領域の欠失と増幅、 さらに、 染色体欠失を伴わないLOHの検出が可能である点で優れている。
このコピー数正常LOHは相同染色体間の組換えと染色体の異常分配によって生じる異常で、 染色体全体あるいはその一部が片親に由来するため片親性二倍体 (uniparental disomy : UPD) と称される。 AAにおけるSNP-Aによる解析は、 当初、 MDSとの鑑別を目的として、 G分染法において 「No growth」 などの結果により情報が得られない場合に有用と考えられていた。 その結果、 最も頻度が高い異常は6番染色体短腕に認められるUPD6pであった (図2)。
図2 HLA-B4002により提示される自己抗原に特異的な細胞傷害性T細胞から、 造血幹細胞および前駆細胞がエスケープする際に推定されるメカニズム
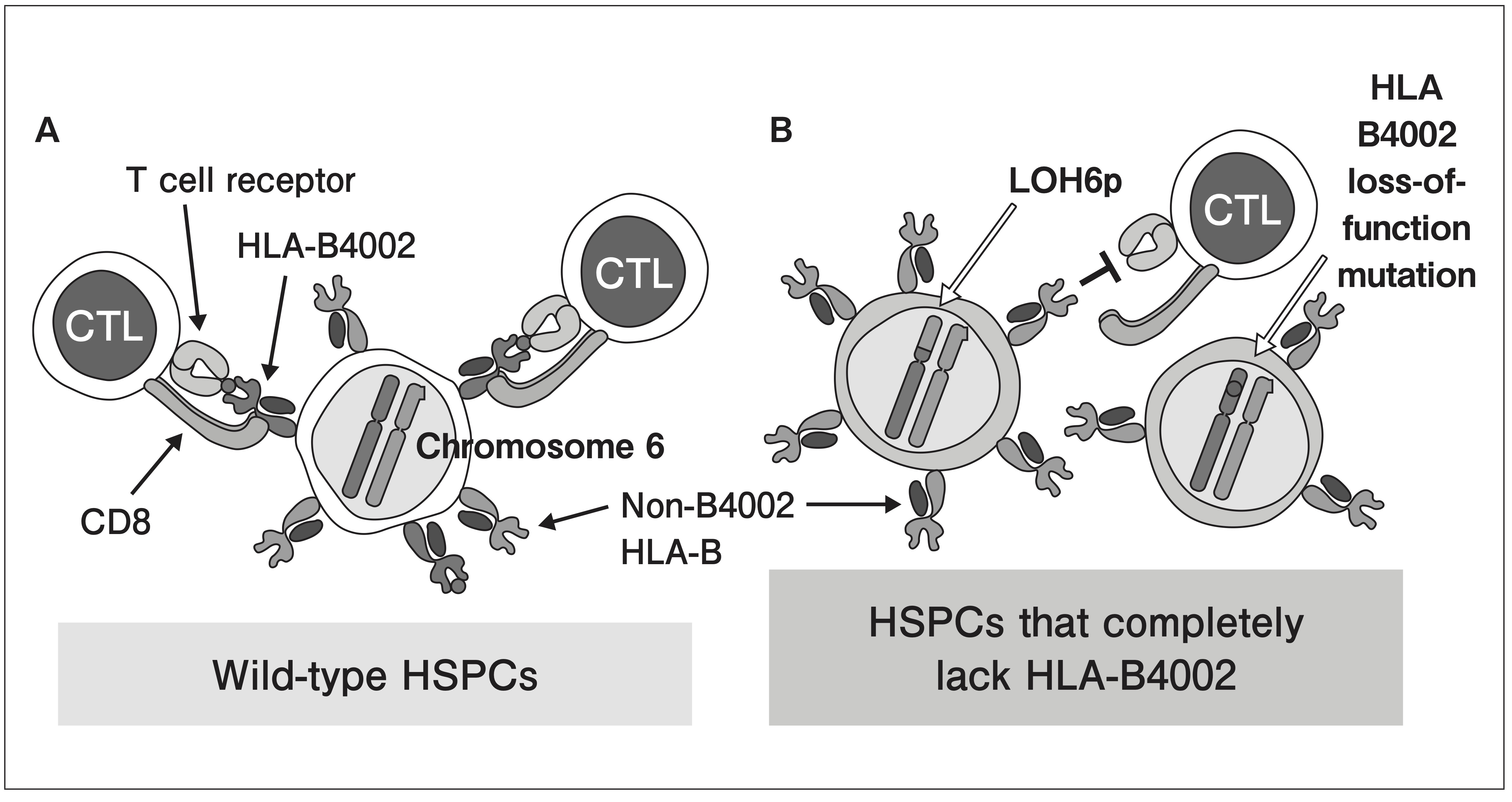
A 正常な造血幹細胞 (HSPCs) は、 HLA-B4002により提示される自己抗原を認識する細胞傷害性T細胞により攻撃される。 しかしながら、 B HLA-B4002にゲノム異常をきたしたHSPCsはこの攻撃からエスケープする。 その機序として、 LOH6pやナンセンス・フレームシフト・スプライス部位の変異によるHLA-B4002の発現欠失が認められる。
〔参考文献34)より引用改変〕
UPD6pは、 G分染法など従来の染色体分析では検出不可能であるが、 SNP-A解析により検出される異常で、 AAの13%程度に認められ²⁰⁾、 この結果は独立したコホートで再現された²⁴⁾。 同一症例で明らかに切断点の異なるUPD6pを有する複数のクローンが存在することから、 AAの患者ではUPD6pを有するクローンが強く選択されていることが示唆される。 注目すべきことに、 これらのUPD6pの領域はHLA class I領域を越えて短腕側で認められることはなく、 UPD6pを示す症例ではHLA遺伝子座は片親由来のhomozygousとなる。
よって、 UPD6pを呈する患者の末梢血では、 Flowcytometryにより、 一方のHLAの発現がタンパク質レベルで消失したクローンが観察された。 この現象は多系統の造血細胞で認められ、 しばしば顆粒球、 単球、 B細胞、 および骨髄のCD34陽性細胞においても確認される。 さらにUPD6pはこれらの分画において年単位で持続することから、 UPD6pを有する造血の起源は幹細胞分画、 おそらく長期の造血を支持する造血幹細胞に生じ、 このクローンが拡大して、 場合によっては末梢血造血のほとんど全てを再構築している可能性が考えられる。
UPD6pのもう一つの大きな特徴は、 同異常を示す症例の多くが、 HLA A*0201、 A*0206、 A*3101ないしB*4002をヘテロ接合で有しており、 UPDではほとんど常にこれらのアレルが消失することである。 したがって、 UPD6pを有するクローン性造血が単に著しく減少した幹細胞に由来することから生じうる、 集団遺伝学でいうところのBottle neck効果で生じているのではなく、 強いDarwin選択による結果である可能性が高いと考えられる。
UPD6pの標的がHLA class I分子であることと、 これに提示される抗原がAAにおけるCTLの標的と考えられていることを考え合わせると、 これらの症例では上記の特定のHLA class I分子上に提示される抗原が免疫学的な攻撃の標的となっており、 UPD6pでは提示に必要なHLAがゲノム上から消失することで、 免疫学的な攻撃からエスケープし、 選択をうけてクローン性造血として観測されると考えられる (図2)。
実際、 この仮説は、 ハプロ一致移植の例においても、 再発時の白血病細胞にUPD6pがしばしば認められ、 その場合、 欠落するHLAアレルは常に移植片対白血病効果の標的となっていると推定される不一致アレルであるという観察結果に合致するものである²⁵⁾²⁶⁾。 AAにおいて高頻度に認められ、 UPD6pにおいて欠失することが明らかとなった上述のHLAアレルは、 データベース上、 日本人を含むアジア人に多いとされている。
欧米のAA例に認められるUPD6pにおいてもこれらのアレルが欠失すること²⁴⁾、 また、 AAと連続した疾患であるPNHにおいても上記のアレルが有意に濃縮していること²⁷⁾、 本症が欧米人と比較してアジア人に多いこと²⁸⁾²⁹⁾ などは、 上記の仮説と併せて、 AAでは、 上記のHLAで提示される特定の抗原を共通の免疫学的標的として自己免疫反応が生じている可能性を示唆するものである。
さらに、 金沢大学からの報告によれば、 UPD6pがあれば、 それ以外のゲノム異常が獲得されなくても長期間にわたってAAにおけるクローン性造血が維持されることが示されており³⁰⁾、 改めてCTLからのエスケープが造血維持に重要であることが確認された。
以上の所見は、 AAの中心的病因が自己免疫機序であることを再確認し、 そのひとつの病態として、 UPD6pによる免疫機序からのエスケープという遺伝学的機序を明らかにすることにつながった。 一方、 興味深いことに、 AAからMDSに進展した症例では、 UPD6pが消失することがあり、 純粋に自己免疫的な病態を呈する症例と、 MDSにより近い症例とを、 UPD6pにより明確に鑑別できる可能性もあり、 今後の詳細な検討が待たれる。
Ⅴ. 再生不良性貧血における体細胞性変異の発見
一方、 近年のゲノムシーケンスにおける技術革新を背景として、 AAにおけるクローン性造血は、 体細胞性変異の観点からも検討されてきた。 まず、 ボストンのDana Faber 癌研究所のLaneらは39例のAA症例において、 造血器腫瘍で変異の報告がある219遺伝子について高深度シーケンスを行い、 9例 (23%) に体細胞性変異を同定した³¹⁾。
ひき続き、ロンドンKing’s CollegeのKulasekararajらは、 150例のAA患者のうち29例 (19%) において32個の体細胞性変異を検出した³²⁾。 変異陽性例での罹病期間 (37ヵ月) は、 変異陰性例 (8ヵ月) と比較して有意に長く、 MDS/AMLへの移行も変異陽性例で11/29 (38%) と変異陰性例6/121 (5.0%) と比較して有意に高頻度であった。 さらには、 変異陽性例ではtelomere長の有意な短小化が認められ、 変異が細胞周期の亢進と関連している可能性が示唆された。
そこでわれわれは、 金沢大学、 Cleveland Clinicのグループと共同し439例のAA症例を標的シーケンスにて解析した。 体細胞性変異はBCOR/BCORL1, PIGA, ASXL1, DNMT3Aに最も高頻度に認められた³³⁾。 BCOR/BCORL1とPIGAの変異は全年齢で等しい頻度で認められ良好な予後と関連する一方で、 ASXL1とDNMT3Aの変異頻度は加齢とともに増加する傾向にあり、 白血病進展と不良な予後に関連していた³³⁾。 しかも、 これら2遺伝子は健常高齢者で認められるARCH/CHIPの原因遺伝子とオーバーラップしていた。
AAにおいて観察される体細胞性変異の存在は、 染色体異常やUPD6pと同様に、 これらの変異を伴ったクローン性造血が存在することを示しており、 遺伝子変異により臨床的に異なる病型を呈した。 また、 AAで変異が認められる遺伝子のうち、 ASXL1, DNMT3AについてはMDS/AMLでも高頻度に変異が観察されるが、 MDS/AMLで高頻度に変異を生ずることが知られているTET2, SF3B1, SRSF2, U2AF1, TP53, IDH1/IDH2, FLT3, NPM1, CEBPA遺伝子の変異はAAでは比較的まれである³¹⁾³²⁾。 また、 BCOR/BCORL1とPIGA遺伝子の変異は、 AAでは高頻度に変異する遺伝子であるが、 MDS/AMLでは他の変異と比較して頻度は少ない。 以上より、 AAはしばしばMDS/AMLへ進展するものの、 両者の変異スペクトラムは、 基本的に異なっている (図3)。
図3 さまざまな造血細胞由来のクローン
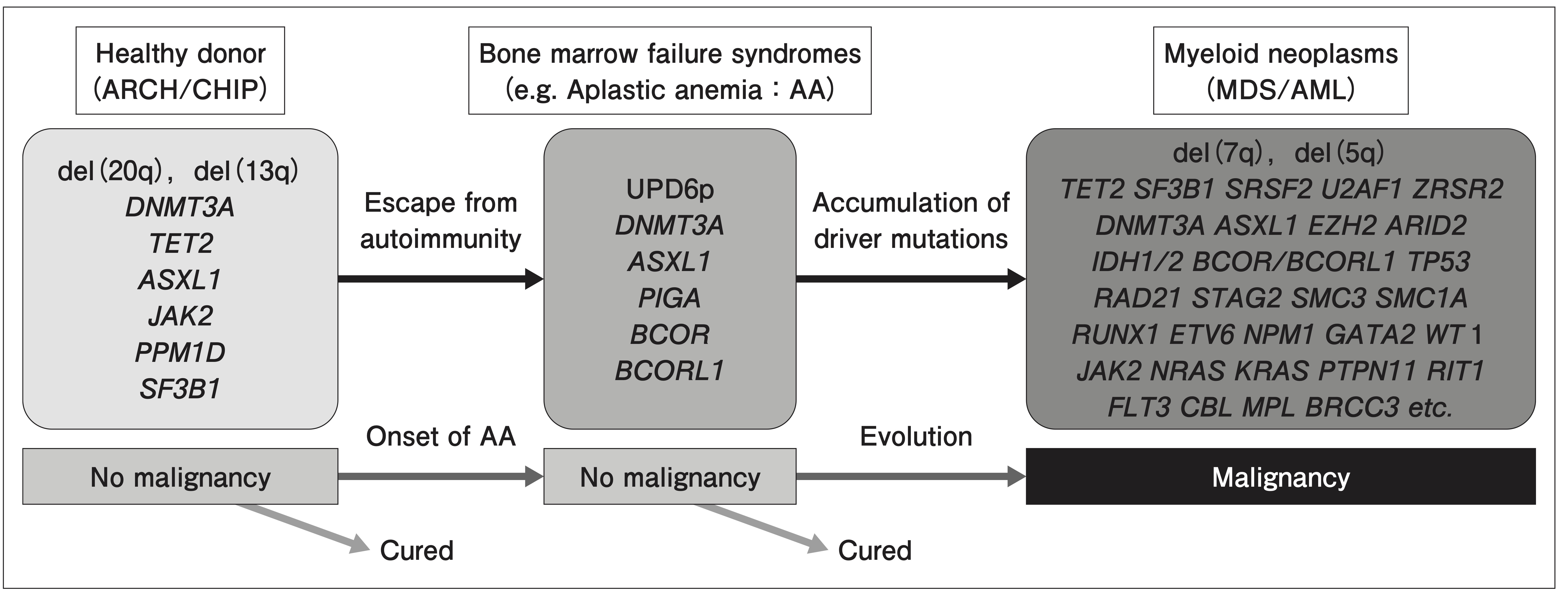
健常者、 再生不良性貧血、 骨髄系腫瘍における造血細胞クローンは臨床経過に伴ってゲノム異常を獲得する。
さらには、 前述のUPD6pに伴う特定のHLA遺伝子座のLOHに関連し、 金沢大学よりAA症例において、 特定のHLA遺伝子座に体細胞性変異が高頻度に報告されている。 ほとんどの変異がナンセンスかフレームシフトをきたし、 そのクローンサイズは病態を説明するほど十分に大きく、 細胞表面へのHLA蛋白の発現が消失していた³⁴⁾。
この所見は、コピー数異常のみならず体細胞性変異により、 AAにおいて認められるクローン性造血がCTLの関与する免疫機構からのエスケープに起因していることを、 示唆している (図3)。 その後の解析により、 最も高頻度に欠失するHLAタイプがB*4002とB*5401であることが明らかとなり、 これらが造血幹細胞抗原をCTLに提示していると考えられている³⁵⁾。
Ⅵ. おわりに
AAと健常人におけるクローン性造血の変異が共通していることから、 AAで認められる変異の一部は、 造血幹細胞にランダムに生じた変異にその起源を有することが推測される。 AAにおいても、 変異の検出される頻度およびその数は、 年齢とともに上昇し、 変異の多くは、 診断時にすでに微小な亜集団として存在していることも明らかにされた。
一方、 両者において高頻度に変異を認めるDNMT3AやASXL1とは対照的に、 健常人や造血器腫瘍で高い割合を占めるTET2 やTP53, JAK2 などの変異はAAでは比較的まれであり、 BCOR, PIGA変異の相対頻度は高くなっている。 このことは、 ランダムに生じた変異が選択される際のメカニズムや骨髄の環境がAAと健常者で異なる可能性を示唆している。 AAにおけるクローン性造血でしばしば認められるPIGA変異やUPD6pについては、 上述したとおり、 造血幹細胞/前駆細胞が免疫学的な破壊からエスケープするメカニズムを指摘している³⁾⁴⁾²⁰⁾。
したがって、 ASXL1, DNMT3A, BCORにおける変異が、 PIGA 変異やUPD6pと同様に、 AAにおける免疫学的異常からのエスケープのメカニズムを担っているという仮説が成り立つ。 こうした点を明らかにするためには、 AAにおける免疫異常の理解、 特に造血幹細胞の傷害に関わるCTLと、 それが認識するエピトープの同定による詳細な病態解明が重要と思われる。
出典
- Young NS, Maciejewski J : The pathophysiology of acquired aplastic anemia. N Engl J Med 336 : 1365-1372, 1997
- Young NS : Acquired aplastic anemia. Ann Intern Med 136 : 534-546, 2002
- Murakami Y, Kosaka H, Maeda Y, et al : Inefficient response of T lymphocytes to glycosylphosphatidylinositol anchor- negative cells : implications for paroxysmal nocturnal hemoglobinuria. Blood 100 : 4116-4122, 2002
- Gargiulo L, Papaioannou M, Sica M, et al : Glycosylphosphati-dylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood 121 : 2753-2761, 2013
- Mathe G, Amiel JL, Schwarzenberg L, et al : Bone marrow graft in man after conditioning by antilymphocytic serum. Br Med J 2 : 131-136, 1970
- Marsh JC, Ball SE, Cavenagh J, et al : Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol 147 : 43-70, 2009
- Scheinberg P, Young NS : How I treat acquired aplastic anemia. Blood 120 : 1185-1196, 2012
- Zoumbos NC, Gascon P, Djeu JY : Circulating activated suppressor T lymphocytes in aplastic anemia. N Engl J Med 312 : 257-265, 1985
- Katagiri T, Qi Z, Ohtake S, et al : GPI-anchored protein-deficient T cells in patients with aplastic anemia and low-risk myelodysplastic syndrome : implications for the immunopathophysiology of bone marrow failure. Eur J Haematol 86 : 226-236, 2011
- Hinterberger W, Adolf G, Aichinger G, et al : Further evidence for lymphokine overproduction in severe aplastic anemia. Blood 72 : 266-272, 1988
- Jerez A, Clemente MJ, Makishima H, et al : STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood 122 : 2453-2459, 2013
- Koskela HL, Eldfors S, Ellonen P, et al : Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366 : 1905-1913, 2012
- Jerez A, Clemente MJ, Makishima H, et al : STAT3 mutaleukemia. Blood 120 : 3048-3057, 2012
- Socie G, Henry-Amar M, Bacigalupo A, et al : Malignant tumors occurring after treatment of aplastic anemia. European Bone Marrow Transplantation-Severe Aplastic Anaemia Working Party. N Engl J Med 329 : 1152-1157, 1993
- Najean Y, Haguenauer O : Long-term (5 to 20 years) Evolution of nongrafted aplastic anemias. The Cooperative Group for the Study of Aplastic and Refractory Anemias. Blood 76 : 2222-2228, 1990
- Tisdale JF, Maciejewski JP, Nunez O : Late complications following treatment for severe aplastic anemia (SAA) with high-dose cyclophosphamide (Cy) : follow-up of a randomized trial. Blood 100 : 4668-4670, 2002
- Dameshek W : Riddle : what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and "hypoplastic" leukemia have in common?. Blood 30 : 251-254, 1967
- Josten KM, Tooze JA, Borthwick-Clarke C, et al : Acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria : studies on clonality. Blood 78 : 3162-3167, 1991
- Griscelli-Bennaceur A, Gluckman E, Scrobohaci ML, et al : Aplastic anemia and paroxysmal nocturnal hemoglobinuria : search for a pathogenetic link. Blood 85 : 1354-1363, 1995
- Katagiri T, Sato-Otsubo A, Kashiwase K, et al : Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood 118 : 6601-6609, 2011
- Negoro E, Nagata Y, Clemente MJ, et al : Origins of myelo-dysplastic syndromes after aplastic anemia. Blood 130 : 1953-1957, 2017
- Mikhailova N, Sessarego M, Fugazza G, et al : Cytogenetic abnormalities in patients with severe aplastic anemia. Haematologica 81 : 418-422, 1996
- Wang H, Chuhjo T, Yasue S, et al : Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome. Blood 100 : 3897-3902, 2002
- Afable MG, Herting C, Lukanowski M, et al : SNP array-based karyotyping : differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood 117 : 6876-6884, 2011
- Vago L, Perna SK, Zanussi M, et al : Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med 361 : 478-488, 2009
- Villalobos IB, Takahashi Y, Akatsuka Y, et al : Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood 115 : 3158-3161, 2010
- Shichishima T, Noji H, Ikeda K, et al : The frequency of HLA class I alleles in Japanese patients with bone marrow failure. Haematologica 91 : 856-857, 2006
- Issaragrisil S, Kaufman DW, Anderson T, et al : The epidemiology of aplastic anemia in Thailand. Blood 107 : 1299-1307, 2006
- Montane E, Ibáñez L, Vidal X, et al : Epidemiology of aplastic anemia : a prospective multicenter study. Haematologica 93 : 518-523, 2008
- Imi T, Katagiri T, Hosomichi K, et al : Sustained clonal hematopoiesis by HLA-lacking hematopoietic stem cells without driver mutations in aplastic anemia. Blood Adv 2 : 1000-1012, 2018
- Lane AA, Odejide O, Kopp N, et al : Low frequency clonal mutations recoverable by deep sequencing in patients with aplastic anemia. Leukemia 27 : 968-971, 2013
- Kulasekararaj AG, Jiang J, Smith AE, et al : Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood 124 : 2698-2704, 2014
- Yoshizato T, Dumitriu B, Hosokawa K, et al : Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med 373 : 35-47, 2015
- Zaimoku Y, Takamatsu H, Hosomichi K, et al : Identification of an HLA class I allele closely involved in the autoantigen presentation in acquired aplastic anemia. Blood 129 : 2908-2916, 2017
- Elbadry MI, Mizumaki H, Hosokawa K, et al : Escape hematopoiesis by HLA-B5401-lacking hematopoietic stem progenitor cells in men with acquired aplastic anemia. Haematologica 104 : e447-e450, 2019


編集・作図:編集部、 監修:所属専門医師。各領域の第一線の専門医が複数在籍。最新トピックに関する独自記事を配信中。

編集・作図:編集部、 監修:所属専門医師。各領域の第一線の専門医が複数在籍。最新トピックに関する独自記事を配信中。